-

蛋白化学成分_蛋白性味_用法与用量_临床应用_蛋白颜色
按水分和固形物所占比重,则含水分87%,固形物13%固形物中大约90%是蛋白质,其中:卵白蛋白75%
-

减肥时蛋白质如何选择
蛋白质不是每天热量的主要来源,却是构成人体器官、荷尔蒙和免疫物质的主要原料,每人每天蛋白质的需要量是
-

蛋白质的作用
蛋白质的作用是什么?蛋白质不仅能帮助你的身体消耗跟多的热,燃烧更多的脂肪,更是塑造肌肉的关键营养元素
-
蛋白质瘦身法原理
蛋白质可抑制促进脂肪形成的荷尔蒙分泌,减少赘肉的产生。最重要的是,蛋白质不会变成无法消失的热能囤积在
-

一天三餐蛋白质减肥食谱
对于很多减肥的人来说,在减肥期间都会刻意的注意蛋白质的吸收,让您重新认识蛋白质,帮助您达到更好的减肥
-

代谢性酸中毒是什么 有哪些治疗方法
当hco3-减少时,h2co3相应增高,离解出co2,使血pco2升高,刺激呼吸中枢,引起呼吸深快,
-

代谢性酸中毒的临床表现有哪些 代谢性酸中毒的发病原因有哪些
对小动脉,一方面因为儿茶酚胺分泌增加使其收缩,另一方面h本身则造成小动脉舒张,严重酸中毒时,后一种作
-

大鱼大肉酸中毒专家教你怎么吃
所以,在旅途中,合理调整饮食结构,多吃一些如豆浆西红柿梨大枣香蕉等润肺生津的食物,不吃或少吃辛辣食品
-
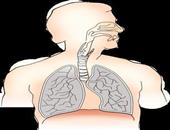
呼吸性酸中毒的病因 呼吸性酸中毒该怎么治疗
呼吸性酸中毒的病因是什么?呼吸中枢抑制药物可以导致肺部功能减弱,从而引发呼吸性酸中毒,常见的药物有各
-

轻度代谢性酸中毒怎么办 如何诊断这个病
如果透析条件尚未具备,可以置胃管持续性抽吸胃酸,一方面可暂时减轻酸血症,另一方面可以吸去体液,为减轻
-

常喝果汁容易让宝宝酸中毒
结果,最近几天突然食欲减退,甚至出现呕吐、头晕的症状,妈妈忙带他去医院,大夫的诊断是,很可能由于乐乐
-

早孕反应一样性别一样吗 胎儿性别怎么鉴定
另外,剧吐还可能会导致人体的水电解质平衡失调,影响肝功能,严重的还可能发生脱水或酮中毒,要是不及时治
-

孕期饮食影响胎儿性别缺乏依据
“谷类食物决定胎儿性别”吗?一些地区在民间盛传某些食物组成的“秘方”可以保证“望子成真”,非洲一些原